- The paper introduces Polyformer, a unified generative model that simulates thermodynamic ensembles from primary sequence and temperature input.
- It employs a novel Diffusion Transformer architecture with ESM-2 embeddings and geometric encoding to capture temperature-dependent conformational transitions.
- Experimental results show close alignment with MD simulations, accurately reproducing folding, disorder, and denaturation trends in protein domains.
Motivation and Problem Setting
Polymeric biomolecules, such as proteins, nucleic acids, and lipids, exhibit dynamic conformational ensembles rather than static structures; their biological functionality is determined by the statistical properties of these ensembles as a function of thermodynamic variables. Existing generative models like AlphaFold solve only the sequence-to-single-conformation mapping, while ensemble and thermodynamics-aware models (e.g., Boltzmann generators, aSAMt) cover either ensemble sampling or temperature dependence, but not both with folding capabilities. Polyformer provides a unified solution: it samples the thermodynamic ensemble of conformations, including sequence-dependent folding and temperature-induced denaturation, for polymeric molecules given only primary sequence and thermodynamic parameters.
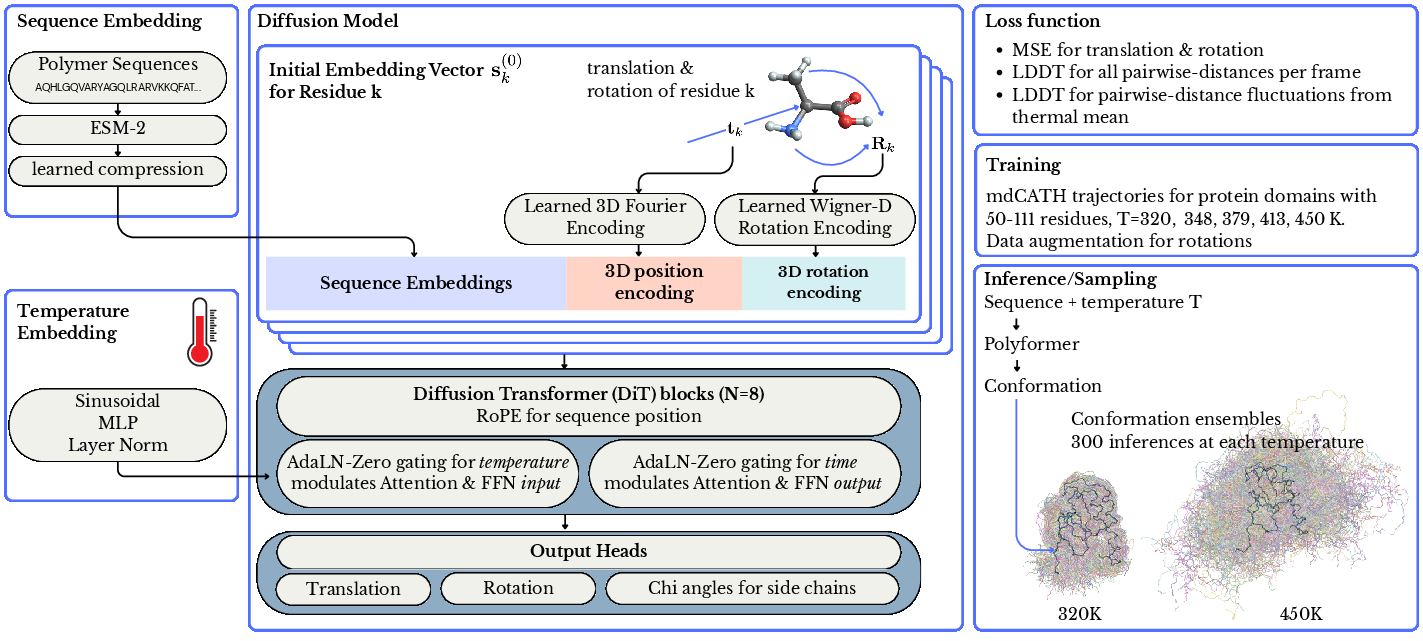
Figure 1: Polyformer architecture for protein thermodynamic model, detailing ESM-2 embedding, geometric encoding, temperature and diffusion conditioning, attention bias, and multi-headed outputs.
Model Architecture and Methodology
Polyformer implements a Diffusion Transformer (DiT) architecture conditioned on molecular sequence and temperature. The input encoding concatenates ESM-2 embeddings, geometric translation via learnable 3D Fourier vectors, and rotation via Wigner D-matrix up to ℓmax=2 (34 rotational features per monomer). This enables representation of both local backbone geometry and internal orientation, generalized across monomer types.
Forward diffusion corrupts translations with linear noise schedule and rotations with Isotropic Gaussian on SO(3). Polyformer’s trunk utilizes N=8 DiT blocks, attention modulated by sequence and temperature with chain-proximity bias (power-law decay of monomer interaction strength), and adaptive LayerNorm gating for separating temperature and diffusion influence. Temperature acts on block input, while timestep gating operates on block output, ensuring physically meaningful control of ensemble diversity vs. thermodynamic deformation.
Three output heads reconstruct noisy frames via translation residuals, axis-angle rotation vectors, and dihedral (chi) angles for sidechain degrees of freedom. Loss functions include standard translation, rotation and chi terms, and two Local Distance Difference Test (LDDT) variants: frame-level and ensemble-level, the latter penalizing deviation from MD-derived ensemble means (multi-scale pairwise distances at specified temperatures).
Inference employs reverse diffusion (DDIM) with rotations denoised via geodesic interpolation on SO(3). Polyformer generates 300 conformations per domain per temperature, conditioned on normalized thermodynamic observable.
Empirical Results
Qualitative and quantitative evaluations utilize the mdCATH benchmark set, comprising molecular dynamics trajectories of protein domains at 5 temperatures (320–450K). Polyformer closely recapitulates the structural ensemble properties of tested domains, including denatured, disordered, and ordered states.
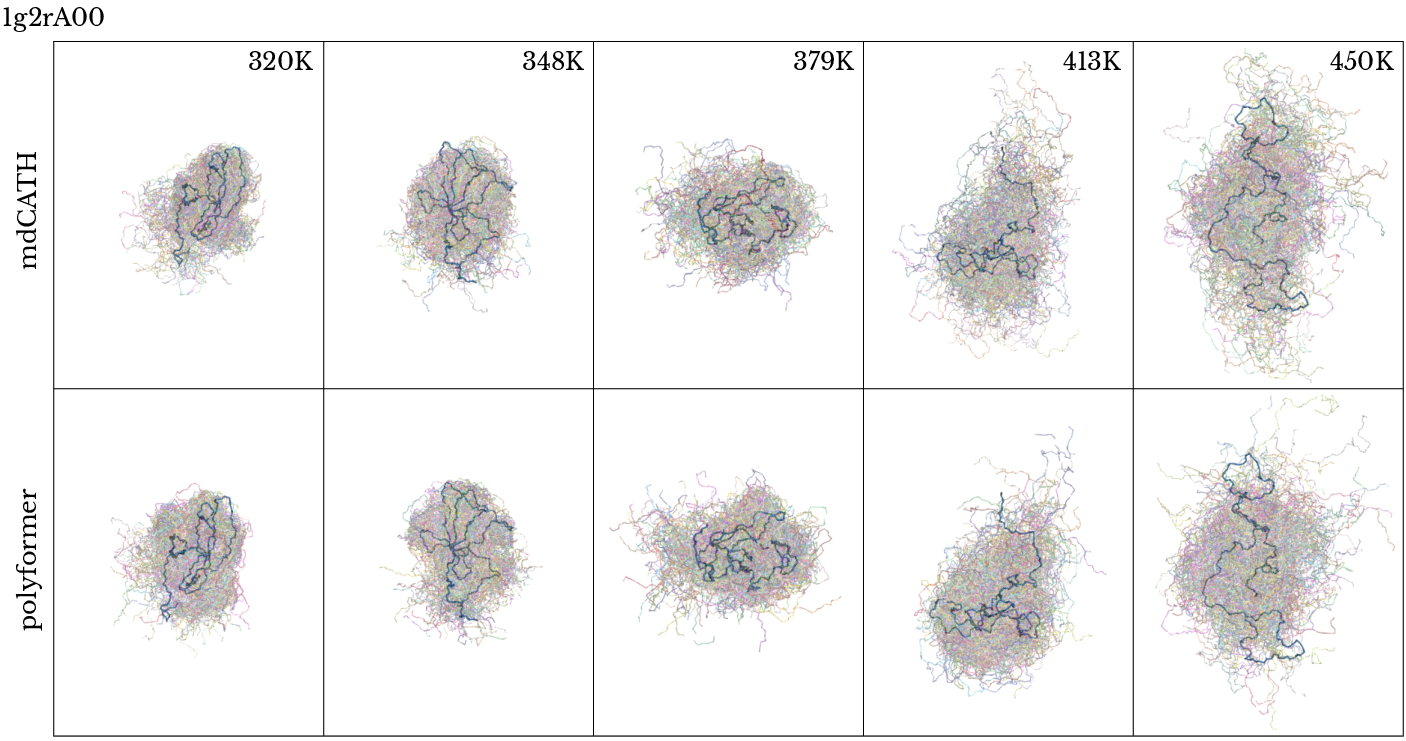
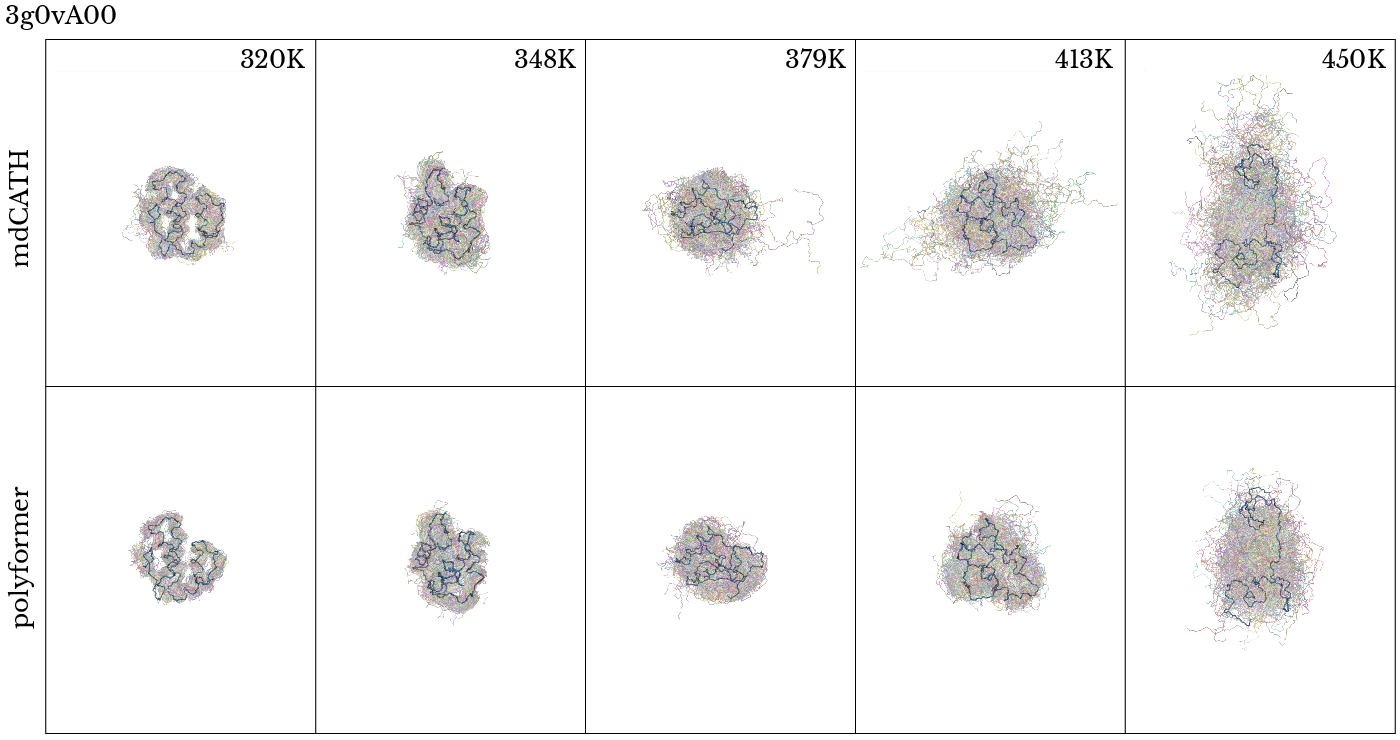
Figure 2: Qualitative comparison of Polyformer and MD-sampled ensemble for lg2rA00 and 3g0vA00; temperature-dependent disorder and radius of gyration trends are preserved.
The backbone dihedral angle distributions (Ramachandran plots) across temperatures exhibit statistically similar shifts in secondary structure populations (α, β) between Polyformer and MD trajectories. Root Mean Square Fluctuations (RMSF) and radius of gyration (Rg) distributions reproduce both the magnitude and temperature dependence of conformational disorder and expansion.
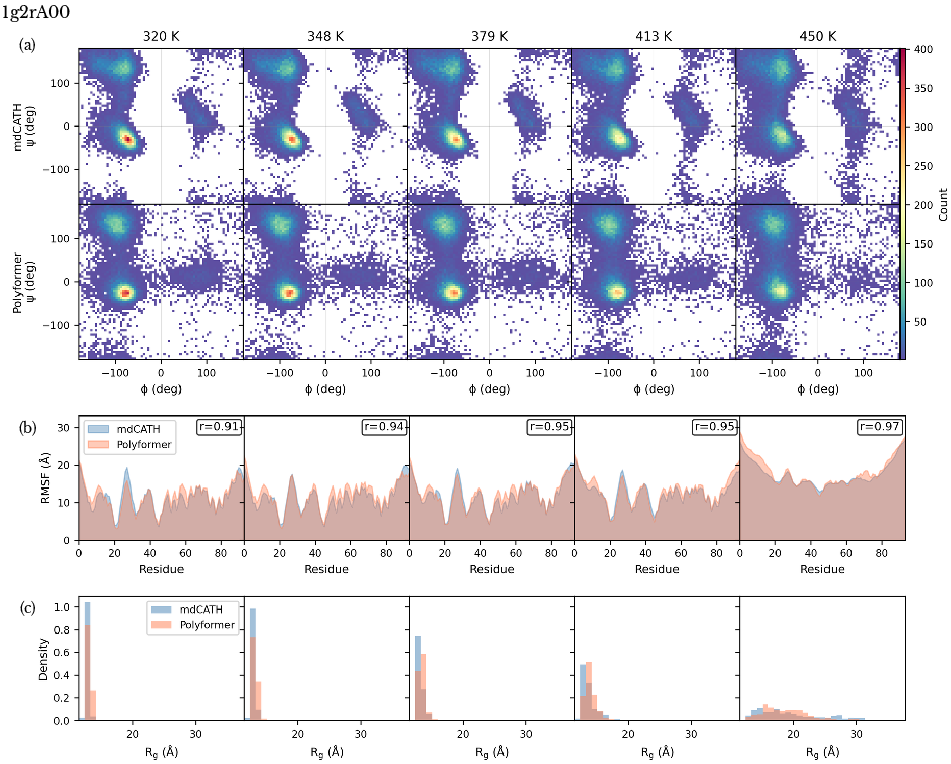
Figure 3: Quantitative comparison for lg2A00: (a) Dihedral angle distributions, (b) RMSF along backbone, (c) Rg distributions as a function of temperature; Polyformer aligns closely with MD reference.
Mean and standard deviation of Rg across sampled ensembles track MD results with high correlation, especially at lower temperatures, with slight systematic deviations at the highest disorder regimes.
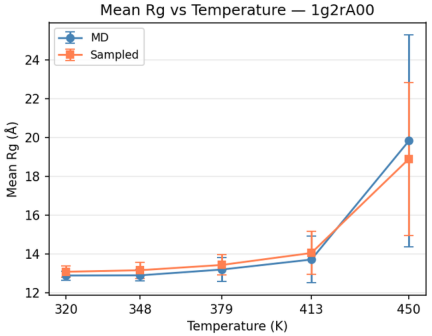
Figure 4: Comparison of mean and standard deviation of Rg for Polyformer vs. mdCATH ensemble sampling, domain lg2A00.
Across all tested domains, ensemble statistics predicted by Polyformer show robust matching to MD, indicating successful generalization of thermodynamic properties from limited training data (2,142 domains).
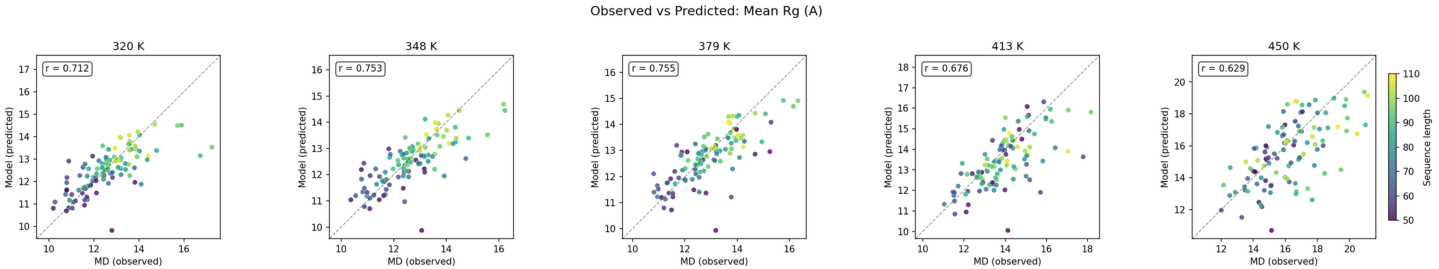
Figure 5: Mean Rg comparison over all test domains and temperatures; correlation between Polyformer predictions and MD trajectory statistics.
Learned Fourier embedding vectors reflect physically meaningful length-scale distributions, aligning with the statistical spectra of Cα–Cα pairwise distances inherent to polymer chains.
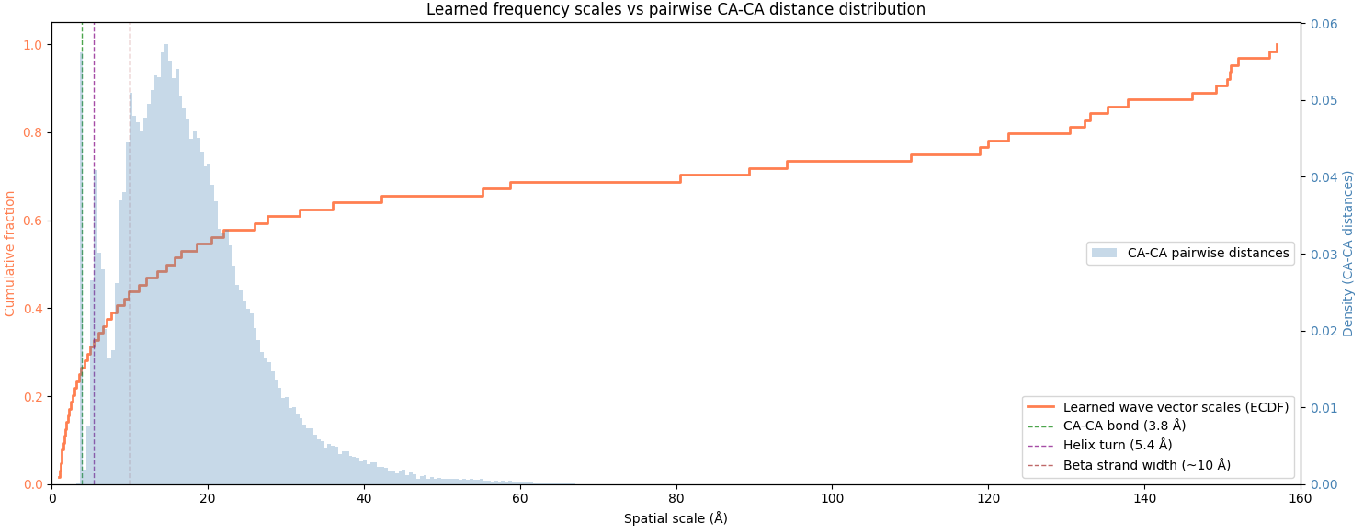
Figure 6: CDF of inverse learned reciprocal vectors (orange) and PDF of Cα0–Cα1 distances (blue) demonstrate model adaptation to polymer physics.
Architectural Decisions and Implications
Polyformer’s modular encoding of monomer positions and local internal degrees of freedom facilitates extensibility to arbitrary polymers. The inclusion of the chain-proximity bias addresses the polymer persistence length and its temperature dependence, critical for physical realism and compositional generality, while the abandonment of Pairformer architecture in favor of learnable Fourier Embedding reduces model complexity and parameter count. Dual-path conditioning for temperature and diffusion enables simultaneous modeling of ensemble diversity and thermodynamic deformation, crucial for physically interpretable generative sampling.
The ensemble LDDT loss serves as a direct supervision signal for temperature-dependent structural changes, encouraging the model to reproduce statistical averages over MD-sampled ensembles, not merely frame-by-frame fidelity. This loss encourages Polyformer to learn free energy landscapes implicitly, since accurate ensemble sampling as a function of temperature relies on proper Boltzmann weighting.
Comparative analysis with previous models reveals that Polyformer uniquely accomplishes all three tasks—folding, ensemble sampling, and thermodynamic dependence—using only sequence and temperature input, without reliance on initial low-temperature conformations or computationally intensive scoring. Its data efficiency is noteworthy: trained on a fraction of the PDB-scale datasets used by AlphaFold/RoseTTAFold, Polyformer demonstrates competitive folding and ensemble prediction, enabled by richer and more informative MD-derived training data.
Limitations, Future Directions, and Theoretical Implications
Current limitations arise from the mdCATH training data: sampling is biased toward smaller, ordered domains, with limited ergodicity at lower temperatures and incomplete coverage of disordered proteins. Improvements in force field accuracy, enhanced ergodic sampling (e.g., replica exchange MD), and broader, more representative ensembles will further improve generalization. Polyformer’s methodology is readily transferable to other polymer classes; monomer encoding can generalize to nucleic acids, synthetic polymers, and mixed systems.
Ablation of ESM-2 embedding contributions and scaling studies will clarify the relationship between sequence encoding and conformational statistics, while advances in attention bias and geometric encoding may enable larger or more complex systems. The implicit learning of free energy landscapes opens the prospect for ML-driven importance sampling and active learning in MD simulations. Polyformer may catalyze rapid iteration in data-driven polymer physics, biophysics, and molecular design, nearing the capability to predict thermodynamic properties and functional conformational ensembles from primary sequence and environmental parameters.
Conclusion
Polyformer achieves unified generative modeling of thermodynamic conformational ensembles for polymeric molecules, capable of sequence-dependent folding, statistical ensemble sampling, and prediction of temperature-induced structural transitions. Its architecture is modular and data efficient, leveraging geometric encodings, chain-physics bias, and dual conditioning to drive high-fidelity ensemble generation from limited MD trajectory data. The framework is extensible to any polymer system, presenting practical and theoretical advances in biomolecular modeling, with implications for machine-learning-driven exploration of free energy landscapes and design of functional polymers.