- The paper demonstrates that orbital-optimized DFT reliably computes CO2 excited-state dissociation curves, reducing excitation energy errors to under 0.5 eV compared to high-level methods.
- It employs complex-valued orbitals to preserve correct spatial symmetry, ensuring accurate characterization of both valence and diffuse Rydberg states.
- The method avoids the pitfalls of active space selection, offering a cost-effective, scalable approach for modeling photochemical processes in small to medium-sized systems.
Valence and Rydberg Excited State Bond Dissociation Curves of CO2 from Orbital-Optimized Density Functional Calculations
Introduction
This work presents a comprehensive assessment of orbital-optimized (OO) density functional theory (DFT) for the calculation of bond dissociation curves in electronic excited states, focusing on benchmark cases in the CO2 molecule: the lowest π∗ valence, $3s$ Rydberg, and 3pσ Rydberg states. The significance of this study stems from the computational limitations of high-level multireference methods for excited states and the well-acknowledged deficiencies of linear-response time-dependent DFT (TD-DFT) when treating states characterized by large electron-density rearrangement, such as Rydberg or charge-transfer excitations. The authors systematically analyze the impact of functional choice, orbital representation (real vs. complex), and basis set completeness on excitation energies and potential energy surfaces, while benchmarking OO-DFT against EOM-CCSD and multireference configuration interaction (MRCI) reference data.
Methodological Advances
The study employs direct orbital optimization via variational techniques targeting energy saddle points, using complex-valued molecular orbitals to retain correct spatial and point-group symmetries for excited states. For single-determinant functionals, spin purification is used to recover pure singlet-triplet energies from spin-mixed OO solutions. The calculation protocol is implemented in GPAW (real space grid and atomic basis) and ORCA (TD-DFT for further comparison).
A critical methodological aspect concerns the necessity of complex orbitals in correctly describing π→3s and π→π∗ excitations in linear molecules. The OO approach enables direct, unconstrained optimization of excited-state Slater determinants, avoiding typical tracking and non-convergence issues of conventional ΔSCF or linear-response approaches.
Proper representation of Rydberg states, whose orbital density is highly diffuse, is evaluated by comparing spatial orbital densities from grid and basis set calculations.
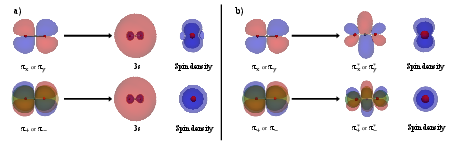
Figure 1: Complex orbitals preserve the cylindrical symmetry of π→3s and π→π∗ excitations in CO20, which is broken when using real orbitals.
Basis Set Evaluation and Orbital Representation
A direct comparison is performed between real space grid, aug-cc-pVDZ, and d-aug-cc-pVDZ basis sets for CO21 Rydberg states at the OO/PBE level.
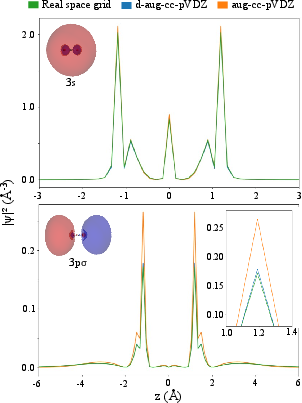
Figure 2: Rydberg orbital densities calculated with d-aug-cc-pVDZ closely match real space grids; aug-cc-pVDZ is insufficiently diffuse for 22.
Results demonstrate that d-aug-cc-pVDZ achieves good agreement with real space grid results for both 23 and 24, while the aug-cc-pVDZ basis is qualitatively inadequate for Rydberg states due to excessive confinement of the orbital densities. This underscores the importance of using sufficiently diffuse functions for quantitative excited state calculations.
Excitation Energies and Functional Dependence
A systematic comparison is performed for vertical excitation energies of the 25 and 26 states using both TD-DFT and OO-DFT with a range of functionals (PBE, B3LYP, PBE0, BHHLYP, CAM-B3LYP). High-level EOM-CCSD reference values from the literature are used as benchmarks.
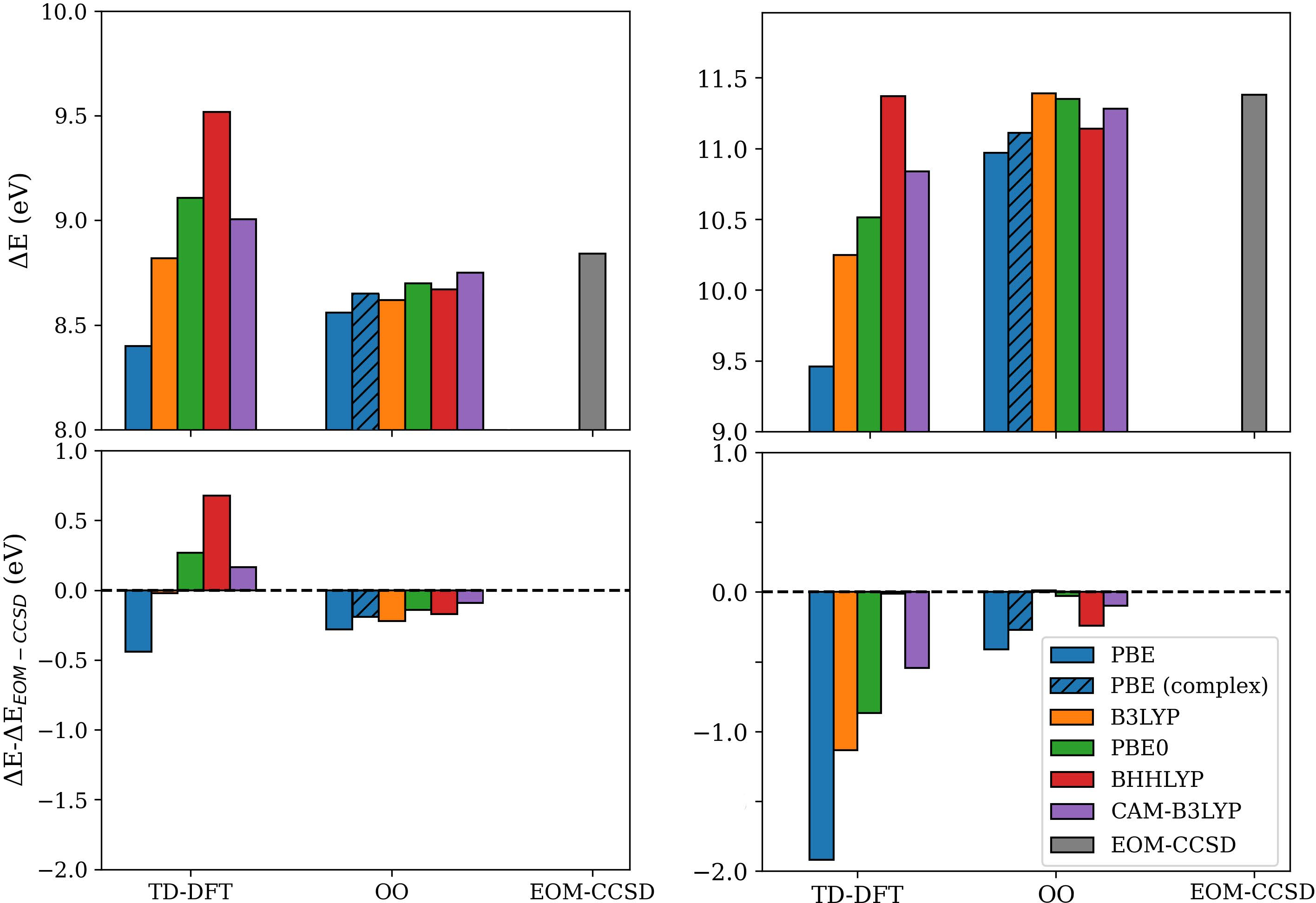
Figure 3: Excitation energies for 27 (left) and 28 (right) Rydberg states from OO and TD-DFT are compared to EOM-CCSD references; OO energies show reduced functional dependence and improved agreement.
Key findings:
- TD-DFT severely underestimates excitation energies for Rydberg states, especially for the more diffuse 29 (π∗0 eV error with PBE).
- For OO-DFT, errors relative to EOM-CCSD are always π∗1 eV, and the sensitivity to functional choice is strongly reduced.
- Using complex orbitals in OO calculations systematically improves accuracy for degenerate states due to the correct symmetry, as shown in Figure 1.
- Hybrid and range-separated functionals further reduce excitation energy errors in OO-DFT.
Potential Energy Surfaces of Excited States
The C–O bond dissociation curves are computed for ground and excited states (π∗2, π∗3, π∗4) using OO-DFT/PBE with complex orbitals.
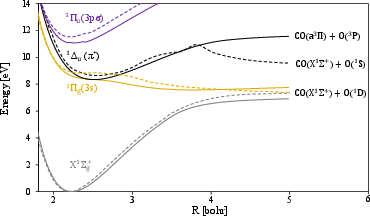
Figure 4: C–O dissociation energy profiles for ground, π∗5, π∗6, and π∗7 excited states, compared to EOM-CCSD/MRCI reference curves.
The OO-DFT potential energy curves:
- Reproduce the overall shapes and energetic orderings of state crossings (notably π∗8/π∗9 double crossing) observed in high-level reference data.
- Undervalue absolute excitation energies by an approximately constant offset, facilitating systematic error compensation in dynamical simulations.
- Provide an accurate description of both valence ($3s$0) and highly diffuse Rydberg states using a single low-cost theory level.
Exceptions arise in bond-elongated limits for specific states; e.g., deviations in $3s$1 dissociation are observed, which may be attributable to inconsistencies in the adiabatic vs. diabatic nature of the reference surfaces or limitations of the single-determinant approach.
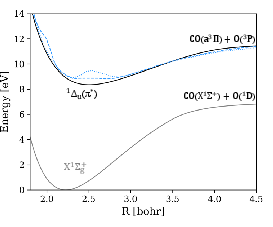
Figure 5: The OO/PBE energy curve of the $3s$2 state aligns with MRCI reference curves except near the $3s$3 crossing.
Importantly, OO-DFT does not rely on active space selection, in contrast to multireference methods, thereby avoiding associated artifacts such as artificial bumps and overestimated barriers in the energy surface.
Discussion and Implications
The results demonstrate that OO-DFT is competitive with, and in several respects superior to, TD-DFT for quantitative modeling of electronically excited states in small and medium-sized molecules, particularly in scenarios involving Rydberg and other "difficult" excitations. The protocol's robustness against choice of functional, basis set, and excited state character strongly facilitates the construction of multi-dimensional potential energy surfaces for nonadiabatic and photochemical simulations.
The intrinsic computational efficiency of OO-DFT opens practical perspectives for application to condensed phase and surface systems, including CO$3s$4 clusters and ices, which are of high importance in astrochemical models and materials-driven photochemistry. Extension to periodic boundary conditions and large-scale systems is straightforward within the GPAW implementation.
Limitations remain due to the single-determinant nature of KS-DFT and the self-interaction error, which can occasion overdelocalization in weakly-bound or highly delocalized excited states (e.g., molecular clusters or Rydberg manifolds). Explicit self-interaction corrections and multi-determinantal approaches may mitigate these effects in future developments.
Conclusion
This work validates OO-DFT as a viable and accurate alternative to high-level correlated wavefunction techniques for the calculation of excited state dissociation curves, particularly in the context of photophysical and photochemical processes involving both valence and Rydberg excitations. By employing complex orbitals and a direct variational approach, robust potential energy surfaces can be obtained with errors in excitation energies systematically below $3s$5 eV, a substantial improvement over linear-response TD-DFT. The approach is computationally efficient and scalable, positioning it as a promising tool for theoretical modeling of excited state dynamics in both molecular and condensed phase environments. Future studies may focus on applying self-interaction corrections and investigating extension to cluster and solid-state systems where electronic delocalization is more pronounced.